


PHACILITATE with Dark Horse Consulting
(An excellent place to work after you leave the FDA, btw)

After my recent Red Cross post, a subscriber posted a link to a company, PHACILITATE, that connects “therapy partners” to peeps in the industry.

Therapy partners like, you know, the Red Cross.
PHACILITATE is all about strategic partnering.
Join Us in Building Meaningful Partnerships
As clinical trials, regulations and new technologies constantly evolve, the path to market for biotech can be challenging. We believe that strategic partnerships are key to success at every stage of the clinical development process, bringing valuable insights and expertise to overcome complex obstacles.
I do love a good strategic pairing. Like a nice premier cru Sauternes (2018 Carmes de Rieussec) with Captain Crunch or Frosted Flakes.
Whilst perusing PHACILITATE’s extensive “menu”, I ran across Dark Horse Consulting. This caught my attention, because Dark Horses seem to be all the rage these days. Well, horses in general... Dark ones, Sky ones, Lusitanos…

I took a look at the previously employed by the FDA Dark Horse Consulting Team.
This is Dr. Don Fink. What’s he all about?

Prior to joining Dark Horse Consulting Group, Don provided 33+ years of uninterrupted service to the United States Federal government, first as a Pharmacology Research Associate Fellow at the National Institutes of Health followed by over 27 years at the FDA Center for Biologics Evaluation and Research (CBER). While at FDA/CBER, Don served as the CMC reviewer for over 100 files submitted by Sponsors that included INDs, IDEs, 510(k)s, BLAs and a PMA for products ranging from recombinant therapeutic proteins, biologic cellular therapies, and cellular combination products.
During his time at Dark Horse, Dr. Fink has worked on a myriad of client projects, successfully enabling clients to benefit from early engagement interactions with FDA as well as achieve their product development goals related to First-in-Human IND filings and biologics license application submissions. He finds it particularly gratifying when he can help clients find solutions to what may otherwise seem to be insoluble problems.
Before DHC, Don was an Expert Biologist in FDA/CBER’s Office of Tissues and Advanced Therapies (OTAT), Division of Cellular and Gene Therapies, Cell Therapy Branch where he was engaged in regulatory activities pertaining to investigational cellular products comprised or derived from starting populations of stem/progenitor cells. Don was responsible for CMC oversight of an extensive portfolio of applications that included hematopoietic, mesenchymal, cord blood, placenta, and pluripotent (hESC and induced pluripotent)-derived stem cell therapeutic products.
During his distinguished FDA career, Don organized an FDA advisory committee to address what at the time was the nascent topic of cellular replacement therapies for neurological disorders. The meeting had, as its focus, stem cell-based investigational products. He was also involved with the planning committee for an NIH/FDA co-sponsored workshop highlighting pluripotent stem cell (PSC)-based products in clinical translation. Don established and served as coordinator for an interest group that monitors development of investigational cellular products derived from PSCs giving attention to evolving issues of concern such as the accumulation of mutations during cellular replication that could impact the product safety profile. Don has served as FDA liaison to both the NIH Stem Cell Task Force and the International Society for Stem Cell Research (ISSCR) Task Force for Clinical Translation of Stem Cells. In 2002 Don co-founded and has co-chaired (through 2020) an FDA-NIH interagency working group in partnership with extramural program officers from the National Institute of Neurological Disorders and Stroke that promotes cross-agency dialogue to facilitate clinical translation of cellular and gene transfer-based products.
In addition to his skills as an FDA reviewer of cellular-based products, Don is recognized for his communication capability and knowledge sharing. He has trained and mentored FDA professional staff as well as served on the steering committee and as a faculty member for FDA’s Clinical Investigator Training Course which provides external stakeholders with in-depth information concerning FDA regulations, ethical considerations, and scientific principles that focus on what is important when conducting clinical trials and preparing submissions for FDA review. Don has given numerous outreach presentations and authored or co-authored several book chapters that outline FDA’s approach for the evaluation of stem cell-based therapies.
Man, Dan is like an FDA MEGA-EXPERT. A MegXpert. He trained the FDA staff.
I wonder if he still knows anybody over there. Like I wonder, after 27 years at the FDA, if he still has any connections over there that might be helpful for strategic partners using Dark Horse Consulting services…🤔
Probably not. Anywho…Don really likes helping with First-In-Human IND filings. What’s that, you ask?
IND = Investigational New Drug (IND)
Here’s some info on IND Application from the FDA:
Current Federal law requires that a drug be the subject of an approved marketing application before it is transported or distributed across state lines. Because a sponsor will probably want to ship the investigational drug to clinical investigators in many states, it must seek an exemption from that legal requirement. The IND is the means through which the sponsor technically obtains this exemption from the FDA.
There are three IND types:
An Investigator IND is submitted by a physician who both initiates and conducts an investigation, and under whose immediate direction the investigational drug is administered or dispensed. A physician might submit a research IND to propose studying an unapproved drug, or an approved product for a new indication or in a new patient population.
Emergency Use IND allows the FDA to authorize use of an experimental drug in an emergency situation that does not allow time for submission of an IND in accordance with 21CFR , Sec. 312.23 or Sec. 312.20. It is also used for patients who do not meet the criteria of an existing study protocol, or if an approved study protocol does not exist.
Treatment IND is submitted for experimental drugs showing promise in clinical testing for serious or immediately life-threatening conditions while the final clinical work is conducted and the FDA review takes place.
Emergency Use of an Investigational Drug or Biologic
The emergency use of an unapproved investigational drug or biologic requires an IND. If the intended subject does not meet the criteria of an existing study protocol, or if an approved study protocol does not exist, the usual procedure is to contact the manufacturer and determine if the drug or biologic can be made available for the emergency use under the company's IND.
The need for an investigational drug or biologic may arise in an emergency situation that does not allow time for submission of an IND. In such a case, FDA may authorize shipment of the test article in advance of the IND submission. Requests for such authorization may be made by telephone or other rapid communication means [21 CFR 312.310(d)].
I LOVE it when they slash those pesky regulations for an investigational new drug during a fake an emergency.
→ I think now’s probably a good time for me to to add a friendly reminder that we are still under a slew of EUAs. For Covid and Ebola, among others.
I wrote about this:


Back to Dark FDA Horse Consulting…
I bet a super FDA MegXpert with lots of friends experience at the FDA could be mighty handy in getting Emergency Use INDs pushed through.
Hypothetically, of course.
Don’s great, but who else is at Dark Horse Consulting? Let’s take a look.
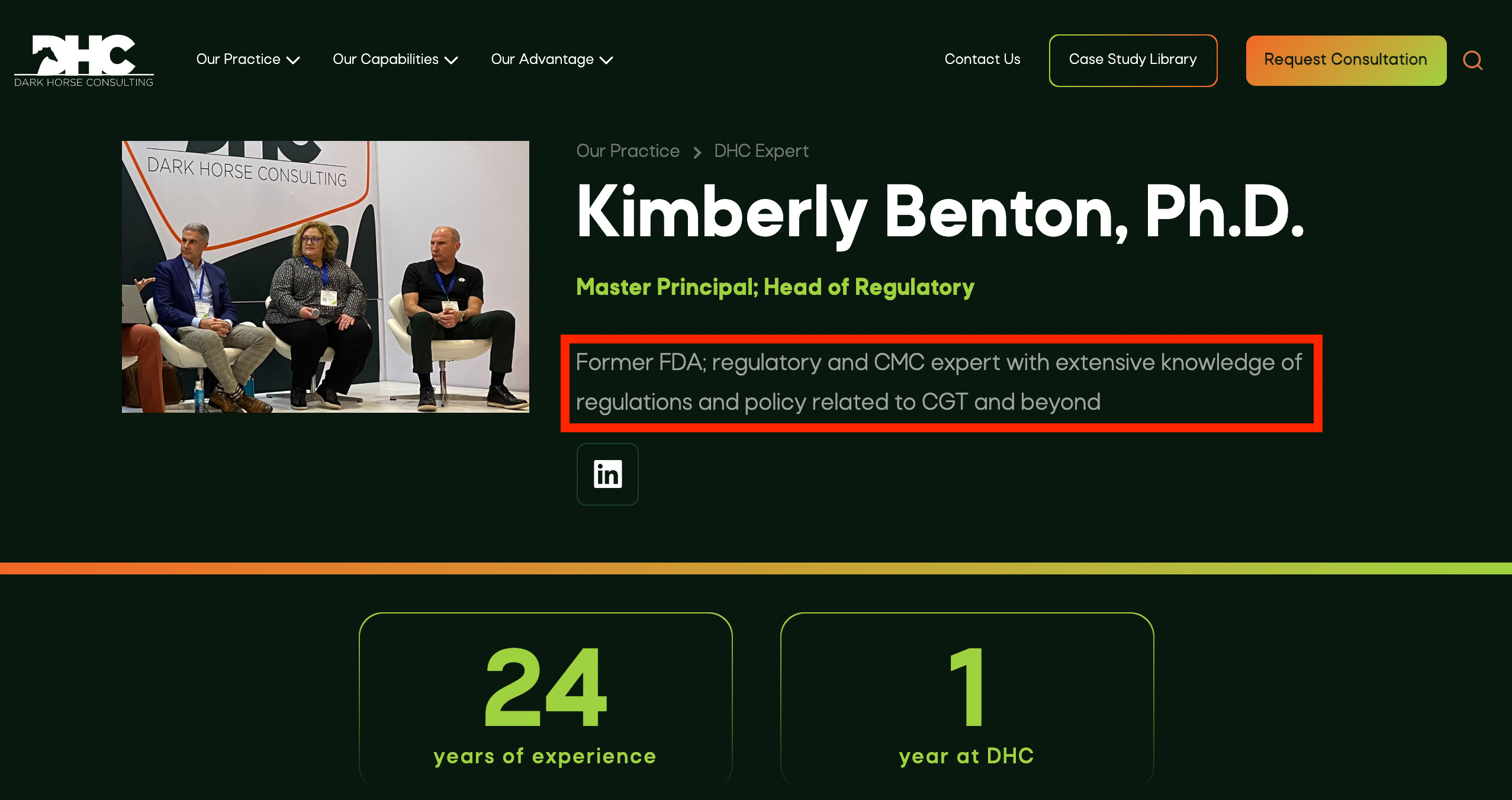
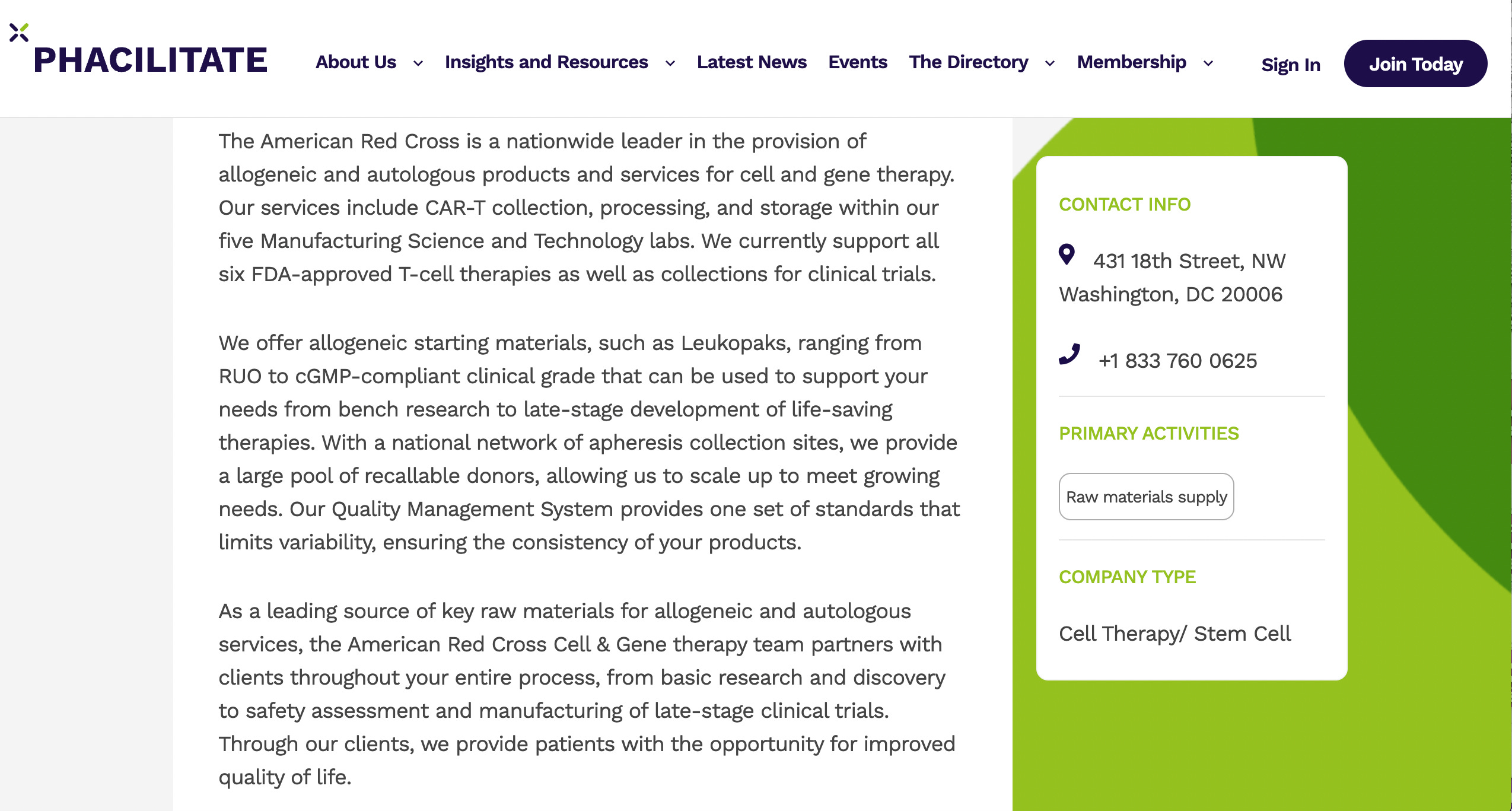
Dr. Benton specialized in cell and gene therapy products during her multi-decade career at the US Food and Drug Administration, Center for Biologics Evaluation and Research (CBER).
Most recently, she served for as Associate Director for Regulatory Management in the Office of Tissues and Advanced Therapies (OTAT). In this role, she directed the regulatory review program for the broad portfolio of products under OTAT’s purview: human tissues, cellular therapies, gene therapies, therapeutic vaccines, xenotransplantation products, and purified and recombinant versions of therapeutic proteins for hematology. For the majority of her career at FDA, she directed and managed chemistry, manufacturing, and controls review, serving in prior positions as Deputy Director of the Division of Cellular and Gene Therapies in the Office of Cellular, Tissue, and Gene Therapies, and Chief of the Cell Therapies Branch.
Kim began her regulatory career as a Chemistry, Manufacturing, and Controls Reviewer of cell therapies, and a research Staff Fellow.
In her cumulative experience at CBER, Kim participated in all aspects of the regulatory review and oversight of cell and gene therapy products, from pre-submission meetings (pre-INDs), IND review, BLA review, advisory committees meetings for scientific topics and for license applications, device review (510(k), PMA, HDE), guidance and policy development, and regulation writing, revision, and interpretation.Other highlights of her time at FDA include initiating regulatory convergence and harmonization efforts on cell therapies with global regulatory agencies including foundational efforts for the International Pharmaceutical Regulators Forum and conferences with the Asia Pacific Association of Southeast Asian Nations (ASEAN).
Dr. Benton's work with DHC clients often focuses on interpretation of communications from regulatory authorities, and development of strategies to achieve the client's regulatory goal(s) by ensuring that information presented to the regulatory authority in meetings or regulatory submissions is presented as clearly and effectively as possible.
Get outta town…another FDA Alum! With Global experience to boot. AND she can get dem dere cell and gene therapy goals met by coaching partners through regulatory meetings with her former friends at the FDA. Woohoo!!

Hey look, here’s Heath.
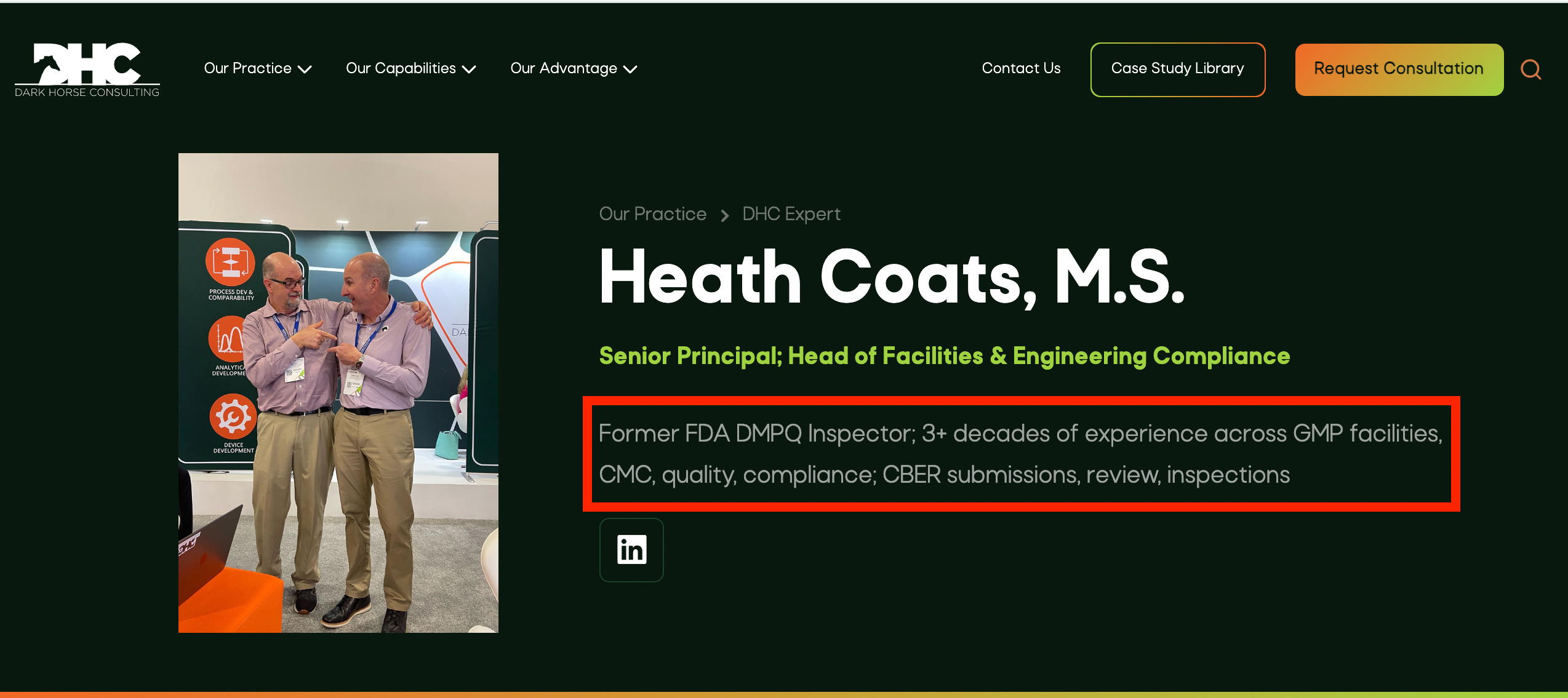
Heath Coats has over 35 years of industry and consulting experience, seven of which were as a Biologist with the Division of Manufacturing and Product Quality (DMPQ) in U.S. FDA's CBER. There, he gained extensive knowledge of administrative and regulatory review procedures for INDs and CMC sections of biologics license applications. During his time at the Agency, Heath reviewed applications and supplements for cell therapy products, HPC cord blood, plasma fractionated products, vaccines, allergenics, aseptic processing, and in vitro diagnostics.
SHUT THE FRONT DOOR?!?! Another FDA Alum!!

For DHC clients, Heath's experience has enabled him to provide support by performing audits and mock audits to support due diligence, vendor qualification, and preparation for regulatory inspection. His extensive inspections history (as delineated in the next paragraph) makes him a natural fit for clients either preparing for inspections or working to remediate processes for future rounds of inspection. He also provides support in the form of regulatory strategy and review.
Prior to his tenure at FDA, Heath was a hands-on manager of the validation program for eight years at a cell therapy contract manufacturing company that also manufactured endotoxin detection products and cell culture media.
→ Fun Fact ⇩

Who even cares about endotoxin testing anyway?
Well, the mRNA market does.
Oh right…that’s because it can accelerate the development and release of herd culling murder shots mRNA-based therapeutics.
You know what I always say...
The gold standard, best practice for dealing with something injectable, that’s never been successful due to all the dying:
RAPID. SPEED.
duh.
Great news. There’s a Manufactured Endotoxin Detection Product expert on staff for your herd culling strategic partnering needs.
Moving on, what’s a consulting group without a viral vector expert anyway?
Oh, hello Scott….
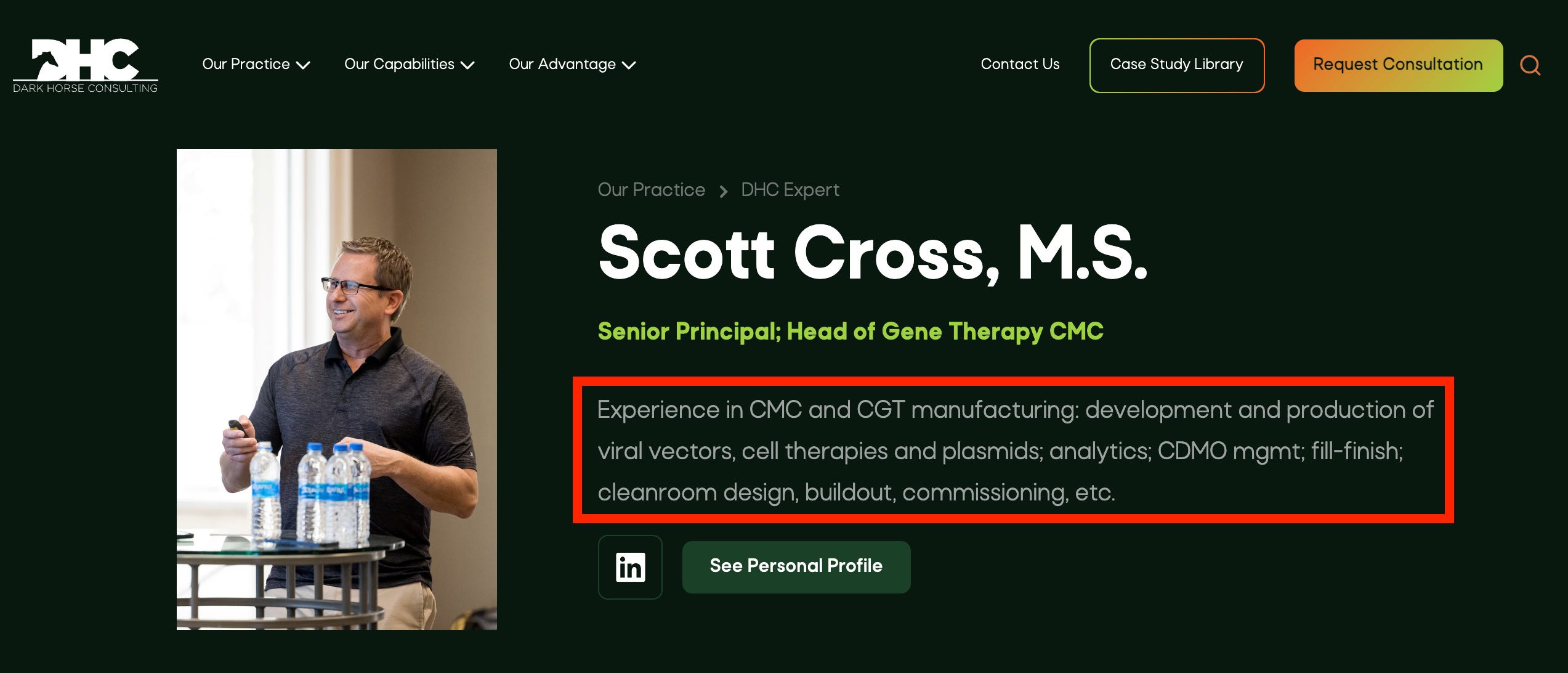
Scott has over 20 years experience working with viral vectors, vaccines and biologics in GMP environments. He has been responsible for cleanroom design and build out and commissioning of vector facilities, as well as the oversight of cell therapy, fill finish and manufacturing support departments.
Scott started his career at Merck and Co. where he worked on the development of an Adenovirus-based HIV vaccine and later, the development, optimization and validation of release assays for live virus vaccines. He then moved to Indiana University (IU) where he managed the IU Vector Production Facility (IUVPF), overseeing the GMP production and testing of Lentiviral and Retroviral vectors for the IUVPF and the National Gene Vector Laboratories (NGVL). While at the IUVPF he also managed the design, build and commissioning of a new viral vector GMP production and testing facility.
After leaving the IUVPF, Scott joined Cincinnati Children’s Hospital Medical Center (CCHMC) managing the GMP Vector Production Facility, Viral Vector Core and the Aseptic Processing Labs. Scott then moved to the University of Florida where he was the Director of Cell Therapy, Fill Finish and Manufacturing Support Operations for Florida Biologix and Brammer Bio. He was also responsible for the design of two new fill finish suites.
Most recently, Scott served as Vice President of Vector Operations at Orchard Therapeutics where he was one of the original ten members and responsible for viral vector development, GMP vector production, plasmid production, oversight of 10 CDMOs, due diligence, and facility design.
🤗 Viral vectors, HIV, Retroviruses, Cell Therapy, Fill Finish, and Plasmid production 🤗 Good GATDAMN….what’s not to love about Scott?!?!?

Meet the Board of Directors:
Ronald loves DNA.
And he’s the EVP of Ancestry.

Ronald Park is a physician with extensive experience in multiple biopharma functions, including drug pricing and reimbursement. A McKinsey & Company alumnus and Stanford MD/MBA, Dr. Park spent sixteen years in various leadership roles at Roche and Genentech, before joining Ancestry as EVP of Health and DNA in 2020. In 2021, Ron became CEO of Ambys Medicines. While at Roche/Genentech, Ron co-founded and co-led Roche Personalized Healthcare Center of Excellence, leading the company’s efforts to globally accelerate the use of genomics, real-world data, and digital health for the development and delivery of medicines to patients with serious diseases.
Don’t even start about Ancestry. They are just telling you about your family and stuff. And your data is totally safe.
And Ronald’s affiliation to Ancestry has NO OVERLAP with Dark Horse Consulting, assholes. I can’t believe you would even think that.
Genealogy websites have become increasingly popular in recent years. These platforms are able to scour the web in search of documents and archival data, which can help users build historically accurate family trees.
There’s also another side to genealogy websites that has attracted attention from privacy advocates: DNA testing. Websites like Ancestry.com can use DNA testing to find matches, but the fact that these platforms store this information on their end means that hackers could try and steal it. Tap or click here to see how Ancestry.com suffered a huge data breach.
Since genealogy websites collect so much data, their user database can be quite valuable in the corporate world. And that’s exactly what’s happening to Ancestry.com thanks to an acquisition by Blackstone — its new parent company. This means if you sent your DNA to Ancestry, Blackstone has it now. Here’s how you can remove it.
Blackstone buys out Ancestry.com
According to new reports from Reuters, the multinational private equity firm Blackstone Group has purchased Ancestry.com for the staggering price of $4.7 billion. This acquisition includes all debt accumulated by Ancestry.com as well, which shows just how eager Blackstone is to add the company into its vast portfolio.
Now that Ancestry.com is under new management, you’re probably wondering what kind of company The Blackstone Group is? Well, for starters, Blackstone deals mostly with private equity, credit and hedge fund investments. Most of its properties are in the financial sector, which makes Ancestry.com a curious purchase altogether.
But if you read between the lines, you can see why the website is so valuable. Ancestry.com is the biggest provider of home DNA testing services, which users can apply towards finding genealogy data and personalized health information.
Ancestry.com boasts more than 3 million paying customers from around the world, and the DNA data it manages is highly valuable to anyone who would be interested in selling it to, say, pharmaceutical companies or medical data firms. It’s almost a no-brainer that a big hedge-fund company would want a slice of the pie.
Of course, if you submitted DNA information to Ancestry.com, this also means your data is at risk of being sold or traded. No, this isn’t illegal either. Once you give the information to Ancestry.com, it’s theirs to use. The terms and conditions more or less spell this out.
Dark Horse Consulting.

Also…we know people 😉

Totally missed opportunity not calling it Pfacilitate. I bet they’re really shaking their heads now
Thank you for this Sarah.
I can't say I am surprised, but I am nevertheless still fucken pissed off at these parasitic psychopaths.
Pendulation awaits them all.
The phuckers